Vikash K Mishra, Mitali Mishra, Soumya Mishra, Prashant Sahu, Sushil K. Kashaw*
Department of Pharmaceutical Sciences, Dr. H. S. Gour University, Sagar, India
* For Correspondence:
Dr. Sushil K Kashaw,
Assistant Professor,
Department of Pharmaceutical Sciences,
Dr. H. S. Gour Central University,
Sagar, India – Pin – 470003
Phone : +91-9425655720
Email:- sushilkashaw@gmail.com
Abstract
Objective: Febrifugine, a quinazoline alkaloid isolated from Dichroa febrifuga roots, shows powerful antimalarial activity against Plasmodium falciparum. This review aimed to illustrate structural aspects of the naturally occurring febrifugine (1) and isofebrifugine (2) and how these issues were ultimately resolved by chemical synthesis. Materials and methods: Although the use of ferifugine as an antimalarial drug has been precluded because of its severe side effects, its potent antimalarial activity has stimulated medicinal chemists to pursue its derivatives instead, which may provide valuable leads for novel antimalarial drugs. This review summarizes historical isolation studies and the chemistry performed which culminated in the correct structural elucidation of naturally occurring febrifugine and its isomer isofebrifugine. It also includes the range of febrifugine analogues prepared for antimalarial evaluation. Results: The range of analogues of (1) prepared for malarial programmes has been highlighted. It is notable that despite the substantial body of literature surrounding febrifugine and isofebrifugine, the detailed biological mechanism, or mechanisms for their antiprotozoal activity are not completely known. Conclusion: Furthermore, many of the more recent biological studies involving febrifugine have been performed using racemic material, as opposed to single optical isomers.
Keywords: Quinazoline, Febrifugine, isofebrifugine, antimalarial compounds, Plasmodium falciparum
Introduction
Febrifugine is a quinazolinone alkaloid first isolated from the Chinese herb Dichroa febrifuga, but also found in the garden plant Hydrangea (Fig. 1) (McLaughlin & Evans, 2010). For centuries in China, the roots of Dichroa febrifuga Lour. (Chinese name: Cha´ng Shan), a saxifragaceous plant, have been employed against malaria fevers, and no parasite resistant against it has been reported. Later febrifugine (1) and isofebrifugine (2) were isolated as active principles against malaria (Kuehl et al., 1948; Koepfli et al., 1949). It was determined in 1971-72 that, independent of the place of growth, the content of 1 and 2 was 0.02-0.05% in the roots and 0.5-0.7% in the leaves of D. febrifuga.
Febrifugine is likely to act by impairing haemazoin formation required for maturation of the parasite at the trophozoite stage (WHO Report, 2001). In vivo activity of (+)-febrifugine proved comparable to the clinically used drug chloroqine (Murata et al., 1998; Kobayashi et al., 1999; Takaya et al., 1999). Total synthesis and structural modifications of febrifugine have been explored (Takaya et al., 1999; Fishman & Cruickshank, 1970; Chien & Cheng, 1970; Zhu et al. 2006-09; Hirai et al., 2003).
The absolute configurations of 1 and 2 were revised through the asymmetric total synthesis of each stereoisomer of febrifugine by Kobayashi et al. (1999). Isofebrifugine (2) is an isomer of febrifugine (1) which is said to isomerize as given below (Koepfli et al., 1947 & 1948).

The emetic and diarrheal properties (Fishman et al., 1970; Chien et al., 1970) associated with febrifugine (1) has limited its usefulness as a chemotherapeutic agent against ma1aria. Subsequent pre-clinical researches have found that febrifugine possesses adverse side effects. Strong liver toxicity has precluded febrifugine as a clinical drug (De Smet, 1997). This review broadly covers the various approaches adopted for modification in febrifugine (1) and isofebrifugine (2) to lead it a safer, economic and potent antimalarial analogue.
Structural analogues of febrifugine
In the 1970s, the synthesis and evaluation of analogues of febrifugine as potential antimalarial drugs demonstrated that the 4-quinazolinone moiety, the nitrogen atom of the piperidine ring and the hydroxyl group are essential for antimalarial activity (Fishman & Cruickshank, 1970; Chien & Cheng, 1970). It was also demonstrated that the absolute configuration of the compound is important since only modest activity is reported for the unnatural enantiomer (Hewitt et al., 1952). Mono- or disubstitution by certain groups on the quinazoline ring system, particularly at positions 5, 6, and 7, resulted in increased antimalarial activity and, in some cases, also increased the chemotherapeutic index.
The methylenedioxyphenyl group was found frequently in natural products and reported to possess some interesting biological activities. Based on this information Chien & Cheng (1970) prepared some 5,6-, 6,7-, and the 7,8-methylenedioxy derivatives (3) of febrifugine and related compounds. These analogues found to be active against Plasmodium berghei. Their toxicity in mice is much lower than that of febrifugine. The therapeutic indices of these compounds are comparable with those of the parent compound.

Fishman & Cruickshank (1970) prepared 3-[β-keto-ɣ-(3-hydroxy-2-pyridyl)propyl]-4-quinazolone (4a-c), in which the piperidine ring of the side chain has been replaced by pyridine.


Compounds 4a-c were assayed against P. beryhei in mice and Plasmodium yallinaceum in chicks (Osdene et al., 1967), no antimalarial activity was observed. However, the chemical and pharmacological characteristics of 1 encouraged medicinal chemists to pursue suitable lead compounds based on 1 for the development of novel antimalarial drugs. Antimalarial screening demonstrated that febrifugine analogues bearing a modified or unmodified 4-quinazolinone ring are active, while analogues produced through the modification of the side chain attached to the N-3 position of the 4-quinazolinone ring are ineffective. (Fishman & Cruickshank, 1970; Chien & Cheng, 1970; Hewitt et al., 1952) Additionally, an enantiomer of 1 prepared synthetically showed decreased activity. (Kobayashi et al., 1999; Hewitt et al., 1952) These results suggested that the 4-quinazolinone moiety, the nitrogen atom of the piperidine ring, and the hydroxyl groups are necessary for the antimalarial activity and that the absolute configuration of these functional groups plays an important role.
In 1999, Takaya et al. synthesized the bicycle acetone adducts 5 and 6 from the natural alkaloids (+)-1 and (+)-2 via a Mannich-type reaction with acetone in the presence of SiO2.


These bicyclo analogues were evaluated for antimalarial activity against Plasmodium falciparum and in vitro cytotoxicity against mouse mammary FM3A. Both analogues 5 (EC50 = 1.6 x 10-9 M) and 6 (EC50 = 2.8 x 10-9 M) showed antimalarial activity with much higher potency in vitro than chloroquine (EC50 = 1.8 x 10-8 M) against the chloroquine sensitive FCR-3 strain and showed high antimalarial activity against the chloroquine-resistant K1 strain. The analogues were found equally cytotoxic to that of the natural alkaloids against mouse mammary FM3A was. Analogue 5 was observed to be 24 times more potent against the mouse malaria parasite Plasmodium berghei than 6 in their in vivo evaluation.
In 2002, Kikuchi et al. transformed (+)-febrifugine (1) by bis-acetylation to afford 7 which showed significantly lower antimalarial activities (EC50 = 1.6 x 10-9 M) than (+)-febrifugine 1 (EC50 = 7.0 x 10-10 M). Further, (+)-febrifugine 1 was N-protected in the presence of ethyl chloroformate to afford the analogue 8 which also showed diminished antimalarial activities (EC50 = 4.8 x 10-6 M) compared to (+)-febrifugine 1. Subsequently, oxidation of (+)-1 afforded keto-analogue 9 which showed antimalarial activities of EC50 = 2.0 x 10-8 M. Reduction of (+)-1 afforded 10 which showed similar antimalarial activity to ketone analogue. Both of these analogues showed high selectivity for Plasmodium falciparum. Kikuchi et al., in further investigation, cyclized the hydroxyl analogue in the presence of dimethoxymethane lead to a mixture of cyclized compounds 11 and 12. Both these analogues (EC50 = 3.7 x 10-9 M) and (EC50 = 8.6 x 10-9 M) showed high antimalarial activities. Similar types of modification were carried out with (+)-isofebrifugine (2) but analogues derived from showed significantly less activity than those derived from (+)-febrifugine 1. Kikuchi et al. also synthesized racemic analogues 13-16, which exhibited moderate antimalarial activity and were not selective against Plasmodium falciparum.
In 2003, Hirai et al. reported isolation, structure elucidation, and synthesis of metabolic products of febrifugine and isofebrifugine. The antimalarial activity of the metabolites and their analogues was also described.
In 2006, Kikuchi et al. reported the synthesis and biological evaluation of a series of analogues based on modifications carried out on the 4-quinazolinone, linker and piperidine ring. The antimalarial activities of each of these analogues against both the chloroquine-sensitive FCR-3 and chloroquine-resistant K1 Plasmodium falciparum strains were evaluated in vitro. The cytotoxicity against mouse L929 cells of each analogue was also evaluated.
The fluorinated analogue 17, showed higher antimalarial activity than that of (+)-febrifugine (1) but it also found to be more toxic. Analogue 18 showed high in vitro and in vivo antimalarial activity. Kikuchi et al. found complete loss of antimalarial activity in analogue 19 and concluded that a basic nitrogen group within the heteroaromatic portion is essential for antimalarial activity.

Figure 1. Plant species from which febrifugine (1) and isofebrifugine (2) have been isolated
Initially analogues 20-22 were synthesized in which the length of the deoxygenated linker was varied, which resulted in a total loss of antimalarial activity. Analogues 23-25 were then synthesized which modified the piperidine ring of febrifugine. No antimalarial activity was reported for these analogues, suggesting that any modifications to piperidine moiety would result in a total loss of activity. Kikuchi et al. concluded that the piperidine moiety and linker of febrifugine were essential to maintain potent antimalarial activity. This important study revealed that good antimalarial candidates, based on 1, could only be accessed through modifications carried out on the 4-quinazolinone ring.
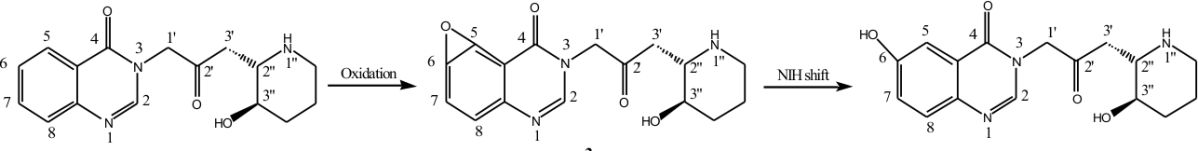
Figure 2 – Metabolic pathway of febrifugine (1)
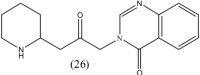
Michael and co-workers in 2006 reported synthesis of racemic deoxyfebrifugine 26. However, no activity has been reported for this analogue.
In 2006, Zhu et al. synthesized and evaluated a range of novel febrifugine analogues based on the work by Hirai et al. Many of the analogues synthesized by Zhu et al. possessed lower toxicity than the parent alkaloid. Based on these findings it was proposed that Febrifugine, 1, is found to be metabolized to the corresponding arene oxide 3 by cytochrome P-450 enzymes (Fig. 2). When arene oxide 3 escapes deactivation process by certain enzymes such as epoxide hydrase or glutathione S-transferase, toxicity can result because this reactive electrophile will form covalent adducts with DNA, RNA, proteins, or other biomolecules of the host. Such binding can cause mutations and result in cell damage. In a recent metabolic study of febrifugine, Oshima and co-workers isolated metabolite (Hirai et. al., 2003). This study indicates that arene oxide is most probably a short-lived reactive metabolic intermediate because metabolite would have been derived from intermediate through the rearrangement known as NIH shift (Guroff et. al., 1967).

It was hypothesized that toxicity of febrifugin can be minimized by blocking the C-5 or C-6 position of the quinazolinone ring or alternatively by increasing the oxidation potential of the molecule. These approaches will make difficult escape of intermediate metabolite and hence might result in lesser toxic compounds. Two analogues 27 and 28 were prepared in this regard which were observed more potent than (+)-febrifugine (1) against both Plasmodium falciparum parasite clones (chloroquine-sensitive) D6 and (chloroquine-resistant) W2. Remarkably both analogues were also found over 100 times less toxic against rat hepatocytes than (+)-1.


In 2009, Zhu et al. designed and synthesized a second set of febrifugine analogues in order to address the isomerization of febrifugine (1) to isofebrifugine (2). In this connection pyrrolidine analogues of febrifugine were synthesized and the antimalarial activity of each was evaluated in vitro against the chloroquine-resistant W2 Plasmodium falciparum strain and in vivo against Plasmodium berghei in mice. Analogues 29 and 30 showed higher antimalarial activity than (+)-febrifugine (1) with lower toxicity. They showed antimalarial activity over four times more potent in vivo. Both were also exhibited 1.3 times less toxic than the existing antimalarial drug chloroquine. The analogues 31 and 32, containing a benzyloxy group on the pyrrolidine ring similarly displayed over 16 times more potent in vitro antimalarial potency than (+)-febrifugine (1) in vivo. However, both these analogues were more toxic than their hydroxyl analogues 29 and 30.
Till the year 2009, Zhu et al., demonstrated that introducing an extra nitrogen atom or an electron withdrawing substitution group on the aromatic ring reduces toxicity while retaining desired biological activity; the original piperidine ring can be replaced by a pyrrolidine ring; the presence of the 3’-methylene group is not necessary for desired antimalarial activity. Based on these findings, Zhu et al., (2012) synthesized some more febrifugine analogues which possess a planar aromatic ring, a 1’’-amino group and C-2’, C-3’’ O-functionality. With these structural features newer analogues are expected to possess same or similar mode of action as of febrifugine. All new compounds were possess a pyrrolidine ring instead of the original piperidine ring and are devoid of the 3’-methylene group. An extra nitrogen atom or a substitution group was introduced on the aromatic ring to block the C-5 or C-6 position of the quinazolinone ring or to increase the oxidation potential of the molecule. Therefore, lower toxicity was expected by reducing or eliminating the tendency to form chemically reactive and toxic intermediates.
New compounds underwent efficacy and toxicity evaluation. Some compounds showed much less toxic than the natural product febrifugine and existing antimalarial drugs and are expected to possess wide therapeutic windows. In Aotus monkeys infected with the chloroquine resistant FVO strain of P. falciparum, one compound 33 exhibited 50% curative dose of 2 mg/kg/day and a 100% curative dose of 8 mg/kg/day. These compounds, as well as the underlying design rationale, were found usefulness in the discovery and development of new antimalarial drugs.
Kikuchi et al., (2014) reported syntheses of quinazoline as well as piperidine ring-modified derivatives of febrifugine. These derivatives were evaluated in vitro for antimalarial activities and cytotoxicities. Compounds were also evaluated in vivo for antimalarial activity. The ring size of piperidine was shown crucial for activity in this study. The tetrahydroquinazoline derivative 34 exhibited potent antimalarial activity with a very high therapeutic selectivity both in vitro and in vivo.
Concluding remarks
In summary, in this review we have aimed to illustrate structural aspects of the naturally occurring febrifugine 1 and isofebrifugine 2 and how these issues were ultimately resolved by chemical synthesis. In addition, the range of analogues of 1 prepared for malarial programmes has been highlighted. It is notable that despite the substantial body of literature surrounding febrifugine and isofebrifugine, the detailed biological mechanism, or mechanisms for their antiprotozoal activity are not completely known. Furthermore, many of the more recent biological studies involving febrifugine (1) have been performed using racemic material, as opposed to single optical isomers.
Conflict of interest
There is no conflict of interest in the present study.
References
Chien P-L, Cheng CC. 1970. Structural Modification of Febrifugine. Some Methylenedioxy Analogs. Journal of Medicinal Chemistry, 13: 867- 870.
De Smet PAGM. 1997. The Role of Plant-Derived Drugs and Herbal Medicines in Healthcare. Drugs, 54: 801-840.
Fishman M, Cruickshank P A. 1970. Febrifugine antimalarial agents. I. Pyridine analogs of febrifugine. Journal of Medicinal Chemistry, 13: 155-156.
Hewitt R I, Wallace W S, Gill E R, Williams J H. 1952. An Antimalarial Alkaloid from Hydrangea. American Journal of Tropical Medicine Hygiene, 1: 768-772.
Hirai S, Kikuchi H, Kim H S, Begum K, Wataya Y, Tasaka H, Miyazawa Y, Yamamoto K, Oshima Y. 2003. Metabolites of febrifugine and its synthetic analogue by mouse liver S9 and their antimalarial activity against Plasmodium malaria parasite. Journal of Medicinal Chemistry, 46: 4351-4359.
Jiang S, Zeng Q, Gettayacamin M, Tungtaeng A, Wannaying S, Lim A, Hansukjariya P, Okunji C O, Zhu S, Fang D. 2005. Antimicrobial Agents and Chemotherapy, 49: 1167.
Kikuchi H, Tasaka H, Hirai S, Takaya Y, Iwabuchi Y, Ooi H, Hatakeyama S, Kim H S, Wataya Y, Oshima Y. 2002. Potent Antimalarial Febrifugine Analogues against the Plasmodium Malaria Parasite. Journal of Medicinal Chemistry, 45: 2563-2570.
Kobayashi S, Ueno M, Suzuki R, Ishitani H, Kim H-S, Wataya Y. 1999. Catalytic Asymmetric Synthesis of Antimalarial Alkaloids Febrifugine and Isofebrifugine and Their Biological Activity. Journal of Organic Chemistry, 64: 6833-6841.
Kobayashi S, Ueno M, Suzuki R, Ishitani H. 1999. Catalytic Asymmetric Synthesis of Febrifugine and Isofebrifugine. Tetrahedron Letters, 2175-2178.
Koepfli J B, Mead J F, Brockman J A Jr. 1949. Alkaloids of Dichroa febrifuga. I. Isolation and Degradation Studies. Journal of American Chemical Society, 71: 1048-1054.
Koepfly J B, Mead J F, Brockman J A Jr. 1947. An alkaloid with high antimalarial activity from Dichroa febrifuga. Journal of American Chemical Society, 69: 1837.
Kuehl F A Jr, Spencer C F, Folkers K. 1948. Alkaloids of Dichroa febrifuga Lour. Journal of American Chemical Society, 70: 2091-2093.
McLaughlin, N P, Evans P. 2010. Dihydroxylation of Vinyl Sulfones: Stereoselective Synthesis of (+)- and (−)-Febrifugine and Halofuginone. The Journal of Organic Chemistry, 75 (2): 518–521.
Murata K, Takano F, Fushiya S, Oshima Y. 1998. Enhancement of NO Production in Activated Macrophages in vivo by an Antimalarial Crude Drug, Dichroa febrifuga, Journal of Natural Product, 61: 729.
Osdene T S, Russell P B, and Rane L. 1967. Journal of Medicinal Chemistry, 10: 431.
Takaya Y, Tasaka H, Chiba T, Uwai K, Tanitsu M, Kim H S, Wataya Y, Miura M, Takeshita M, Oshima Y. 1999. New type of febrifugine analogues, bearing a quinolizidine moiety, show potent antimalarial activity against Plasmodium malaria parasite. Journal of Medicinal Chemistry, 42: 3163-3166.
WHO, WHOSIS, Fact Sheet: Malaria, http://www.who.intlinf-fslen/fact094.html.
WHO, World Malaria Report (2010).
Zhu S, Meng L, Zhang Q, Wei L. 2006. Synthesis and evaluation of febrifugine analogues as potential antimalarial agents. Bioorganic Medicinal Chemistry Letters, 16: 1854-1858.
Zhu S, Wang J, Gudise C, Smith E, Liu X, Zhang Y, Wei L. 2010. Synthesis and evaluation of 4-quinazolinone compounds as potential antimalarial agents. European Journal of Medicinal Chemistry, 1-6.
Zhu S, Zhang Q, Gudise C, Wei L, Smith E, Zeng Y. 2009. Synthesis and biological evaluation of febrifugine analogues as potential antimalarial agents. Bioorganic Medicinal Chemistry, 17: 4496–4502.







